发布时间:所属分类:农业论文浏览:1次
摘 要: 摘要 鳞翅目昆虫, 无论作为农林业主要害虫, 抑或作为传粉媒介和经济昆虫, 对人类社会都产生了深远影响. 昆虫线粒体基因组具有基因组成稳定、基因排列相对保守等性质, 因而广泛用于进化和系统发育研究. 目前已有 63 种鳞翅目昆虫的线粒体基因组序列被测定, 本文比较了其
摘要 鳞翅目昆虫, 无论作为农林业主要害虫, 抑或作为传粉媒介和经济昆虫, 对人类社会都产生了深远影响. 昆虫线粒体基因组具有基因组成稳定、基因排列相对保守等性质, 因而广泛用于进化和系统发育研究. 目前已有 63 种鳞翅目昆虫的线粒体基因组序列被测定, 本文比较了其中已知的 58 条参考序列的基本特征; 同时构建了鳞翅目昆虫系统发育树, 发现其与传统分类具有较好的一致性. 本工作为鳞翅目昆虫的进化研究奠定了基础, 并提示线粒体比较基因组学在后基因组时代对于构建昆虫系统发育关系有着巨大潜力.
关键词鳞翅目线粒体基因组比较基因组进化分子系统学
鳞翅目属于节肢动物门 (Arthropoda) 昆虫纲 (Insecta), 包括蝶(butterfly)、蛾(moth) 2 类, 为全变态昆虫, 1 个世代经历卵、幼虫、蛹、成虫等 4 阶段. 鳞翅目昆虫种类繁多, 世界各地分布约 30 万种(约 95% 以上为蛾类, http://www.discoverlife.org/), 其中已描述的种类有 157424 种[1]. 不论是作为农林业主要害虫, 抑或作为传粉媒介和经济昆虫, 鳞翅目昆虫对人类社会都产生了深远影响. 深入了解鳞翅目昆虫的起源、进化和系统发育关系, 对鳞翅目虫害防治、经济昆虫种质资源的利用具有重要意义[2~5].
动物线粒体基因组(mitochondrial genome)一般为双链、闭环的 DNA 分子[6], 具有突变率高、进化速度快、很少发生重组、易于 PCR 扩增等特点, 已成为一种重要的分子标记[6,7], 并广泛用于包括鳞翅目昆虫在内的许多物种的遗传进化、分类鉴定和系统发育等研究[7~10]. 根据对 NCBI 数据库的统计, 截止到 2012 年 11 月, 完成线粒体基因组测序的鳞翅目物种已达 63 种, 品种数达 112 个(仅次于双翅目的 372 个), 获得参考序列 58 条(为昆虫纲最高). 本文通过比较分析现有的 58 条参考序列, 总结归纳鳞翅目线粒体基因组碱基组成、基因排列、tRNA 和 rRNA 基因、蛋白编码基因、基因重叠区、基因间隔区的特点, 不但有助于了解鳞翅目线粒体基因组的基本特征, 而且可以为高级分类阶元(科、目等)线粒体基因组的比较分析提供参考.
随着国际生命条形码计划(http://ibol.org/)的启动, 线粒体基因 cox1(cytochrome c oxidase subunit 1)作为公认的动物界标准 DNA 条形码基因, 已用于几乎所有动物的分类鉴定等研究[9]. 与 cox1 基因相比, 目前线粒体基因组序列则主要在探讨动物(包括鳞翅目昆虫)的系统发育关系、起源、进化等问题中发挥着重要作用[7~11]. 为此, 本文对线粒体基因组序列在鳞翅目昆虫分子系统学、性别控制以及物种起源与进化等领域的研究进展和应用潜力作一总结与展望.
1 鳞翅目昆虫线粒体基因组的比较分析
1.1 概况
鳞翅目中, 线粒体基因组第一个测序并公布在 NCBI 上的是家蚕(Bombyx mori), 迄今被收录为参考序列的物种包括 29 种蝶类和 29 种蛾类, 分布在鳞翅目蛱蝶科、凤蝶科、灰蝶科、粉蝶科、弄蝶科和蚕蛾科、大蚕蛾科、天蛾科、蝙蝠蛾科、夜蛾科、舟蛾科、灯蛾科、毒蛾科、尺蛾科、潜蛾科、卷叶蛾科、螟蛾科、草螟科等 18 科, 分属凤蝶总科、弄蝶总科、蚕蛾总科、蝙蝠蛾总科、夜蛾总科、尺蛾总科、巢蛾总科、卷叶蛾总科和螟蛾总科等 9 个总科(表 1). 鳞翅目昆虫线粒体基因组大小约 15~16 kb, 平均 15381 bp. 人支蝠蛾[12] 线粒体 DNA(mitochondrial DNA, mtDNA) 由于 A+T富集区较大, 长达 16173 bp, 是已知线粒体基因组最大的鳞翅目物种. 鳞翅目昆虫 mtDNA 碱基组成表现出典型 AT 偏好, 碱基 A+T 含量范围为 77.8%~82.7%, 平均 80.7%. 其中 A+T 含量最小的物种是 O. lunifer, 最大的物种是朝灰蝶.
1.2 基因排列
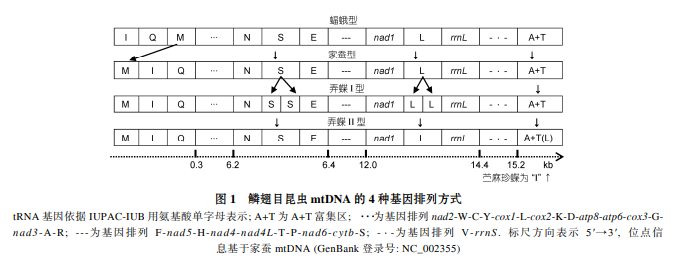
线粒体在好氧真核细胞的细胞代谢过程中发挥着至关重要的作用, 后生动物线粒体基因组的基因组成是高度保守的, 一般包括 37 个基因, 即 13 个蛋白编码基因(protein-coding gene, PCG: NADH dehydrogenase subunits 1-6, nad1-6; cytochrome oxidase subunits 1-3, cox1-3; ATPase subunits 6, 8, atp6, atp8; cytochrome b, cytb), 2个核糖体 RNA基因(rRNA gene: large ribosome RNA, rrnL; small ribosome RNA, rrnS)和 22 个转移 RNA 基因(transfer RNA gene, tRNA gene)[6]. 已知的鳞翅目昆虫线粒体基因组的 13 个蛋白编码基因中 9 个(nad2, cox1, cox2, atp8, atp6, cox3, nad3, nad6, cytb)位于主链(majority-coding strand, J 链 ), 4 个 (nad5, nad4, nad4L, nad1) 位于次链 (minority-coding strand, N链). 除此之外, 还有 1个非编码部分——A+T 富集区, 由于该区域含有许多参与转录、复制的重要调控元件, 因此被认为是 mtDNA 控制区(control region)[43]. 线粒体 DNA 上的基因排列紧凑, 基因间的非编码序列非常短小, 时常可见由少数核苷酸形成的基因重叠[12,16]. 这使得 mtDNA 上的基因排列顺序相对固定[44], 基因重排主要发生在 tRNA 基因[6]. 绝大多数鳞翅目昆虫线粒体基因的排列顺序, 与第一个被测序的鳞翅目昆虫家蚕一致, 而不同于第一个被测序的节肢动物果蝇(Drosophila yakuba) [45]. 例外发生在人支蝠蛾和云南蝠蛾[12]、小赭弄蝶和梳翅弄蝶: 人支蝠蛾、云南蝠蛾线粒体基因组有 22 个 tRNA 基因, 但是排序不同于家蚕(trnMtrnI-trnQ) 而类似果蝇 (trnI-trnQ-trnM) [12,46]; 小赭弄蝶、梳翅弄蝶基因排序类似于家蚕, 却分别有 24 个和 23 个 tRNA 基因——即出现了基因排序异常、基因数目异常两种情形(图 1). tRNA 基因易位在昆虫 mtDNA 进化过程中比较普遍[43,47], 基因重排越明显的物种分子进化速率越快[48], 因此可用于研究物种演化过程. 人支蝠蛾和云南蝠蛾是目前唯一已完成线粒体基因组测序的非双孔亚目(nonditrysia)类群[12], 其特殊的基因顺序或许将为分析鳞翅目高级阶元双孔亚目、非双孔亚目的分歧事件提供参考. 梳翅弄蝶和小赭弄蝶 trnS, trnL 基因个数分别为“3+3”和“2+3”, 不同于常见的“2+2”, 因此颠覆了动物 mtDNA 只有 37 个基因的传统观念[6]. 另外, 梳翅弄蝶额外的 trnS, trnL 基因为串联重复, 归纳为弄蝶Ⅰ型; 小赭弄蝶额外的 trnL 基因位于 A+T 富集区, 归纳为弄蝶Ⅱ型, 朝灰蝶[22]、苎麻珍蝶[19]情形分别与之类似(表 1, 图 1).
1.3 tRNA 与 rRNA 基因
鳞翅目昆虫线粒体基因组中, 22 个 tRNA 基因一般包括[49] 2 个 trnL [trnL(UUR)和 trnL(CUN), trnL1 和 trnL2], 2 个 trnS [trnS(AGN)和 trnS(UCN), trnS1 和 trnS2]以及其他 18 种 tRNA 基因各 1 个. 通常, 对鳞翅目昆虫而言, 除 trnS(AGN)外, 其余 21 个 tRNA 基因二级结构均为典型的三叶草结构(typical cloverleaf secondary structure)(图 2); 而 trnS(AGN)缺少二氢尿嘧啶臂(DHU stem), 形成一个简单的 D 环[14,16] (图 2). 当然也有例外, 如樟蚕的 trnS(AGN), trnS(UCN)二级结构均缺少 DHU 臂[27], 而茶小卷叶蛾[33]等物种的 trnS(AGN)则拥有完整的三叶草结构.
碱基错配(base pair mismatch)普遍存在于鳞翅目昆虫 tRNA 二级结构[14]. 例如, 碱基错配数云南蝠蛾为 18[12], 柞蚕为 24[15], 残锷线蛱蝶为 22[16], 黑纹粉蝶为 19[23], 樟蚕为 24[27], O. lunifer 为 32[35], 美国白蛾为 23[36]. 错配类型包括 G-U, A-C, A-G, A-A 和 U-U 错配, 最常见的是 G-U 错配等. 碱基错配可发生在 tRNA 二级结构的二氢尿嘧啶臂、反密码子臂、 TΨC 臂、氨基酸接受臂, 其中氨基酸接受臂错配频率较高.
rRNA 基因包括 rrnL 基因(16S rRNA 或 lrRNA 基因)和 rrnS 基因(12S rRNA 或 srRNA 基因) [6], 其大小和位置均比较保守, 碱基组成表现出明显 AT 偏好[35]. 其中, rrnL 位于 trnL(CUN)和 trnV 之间, 大小 1311~ 1421 bp, 平均 1350 bp, 其中大紫蛱蝶(S. charonda kuriyamaensis)的长度最小, 黄斑长翅卷叶蛾长度最大. 鳞翅目 rrnL基因的 A+T含量为 81.5%~86.0%, 平均 84.4%, 其中 O. lunifer 最低, 云南蝠蛾最高. 相较 rrnL 而言, 鳞翅目昆虫的 rrnS(位于 trnV 和 A+T 富集区之间 )大小更为保守 [12,14,15,41], 范围为 739~830 bp(不包括欧洲玉米螟的 434 bp 和亚洲玉米螟的 435 bp), 平均 779 bp, 其中祖灰蝶长度最小, 苹掌舟蛾长度最大. 鳞翅目 rrnS 的长度虽然远小于 rrnL, 但是其 A+T 含量却略高于 rrnL[23,29]: rrnS 的 A+T 含量 82.1% ~87.1%, 平均 85.1%, 其中欧洲玉米螟最低, 旋纹潜蛾最高. 一般来说, rRNA 基因的 A+T 含量高于蛋白编码基因和 tRNA 基因[15].
1.4 蛋白编码基因
PCG 的碱基组成表现出明显 AT 偏好[15,28,32,37], 且不同鳞翅目物种的 PCG 数目、排列顺序完全一致, PCG 大小在不同物种中相差不大(图 1). PCG 没有内含子, 这与整个 mtDNA 表现的“非编码区稀少”特征类似, 可能与线粒体进化有关[6,50]. PCG 最常见的起始密码子为 AUN[12], 编码 Met. cox1 基因起始密码子较复杂, 例如, 家蚕为 UUUUAG[13], 亚洲玉米螟、欧洲玉米螟为 UAUUAG[14], 苎麻珍蝶为 UUG[19], 朝灰蝶为 UUAG[22]. 最近一项来自不吉按蚊转录本的证据支持 UCG[51], 而大多数鳞翅目物种[12,16,23,42], 包括来自豆荚螟的表达序列标签数据都支持 CGA. 因此, 认同 CGA 作为 cox1 基因起始密码子的研究占大多数[12]. PCG 终止密码子为 UAA, 某些基因则以不完全的 U 或 UA 作为转录终止信号[13]. 例如, 人支蝠蛾 nad2, cox1, cox2, nad3, nad5 基因为 U, 而其 atp6, nad4L, nad6 基因为 UA; 云南蝠蛾 cox1, cox2, nad3, nad5 基因为 U, 而 atp6, nad4L, nad6 基因为 UA[12]; 野桑蚕和家蚕 cox1, cox2 基因为 U[13]; 亚洲玉米螟和欧洲玉米螟 cox2, atp6 基因分别为 U, UA[14]; 柞蚕 cox1, cox2, nad3, nad5 基因为 U, atp6 基因为 UA[15]; 残锷线蛱蝶 nad4 基因为 U[16]; 黑纹粉蝶 cox1, cox2, nad2, nad5基因为 U, nad3基因为 UAG[23]. 一般认为, 不完全终止子 U, UA 在转录后经聚腺苷酸化(posttranscriptional polyadenylation)形成完整的 UAA 终止子[52,53].
1.5 密码子使用与进化
(1) 密码子使用. 利用 Codon Usage Database数据库[54]收录的 12 种以及未发表的 20 种(表 1), 合计 32 种鳞翅目昆虫 mtDNA(共 110990 个完全密码子), 统计 PCG 密码子使用情况和碱基组成, 分析密码子偏好性. 统计结果经汇总整理见表 2, 从表中可见鳞翅目 PCG 密码子使用存在以下特征: (i) 鳞翅目昆虫有 2 种完全终止密码子 UAA 和 UAG, UAA 使用频率远高于 UAG; (ii) UUA, UUG 和 CUN 等 6 种密码子均对应氨基酸 Leu, UUA 使用频率最高; (iii) UUU 和 UUC 对应氨基酸 Phe, UUU 使用频率更高; (iv) AUU 和 AUC 对应氨基酸 Ile, AUU 使用频率更高; (v) AUA 和 AUG 对应氨基酸 Met, AUA 使用频率更高; (vi) AAU 和 AAC 对应氨基酸 Asn, AAU 使用频率更高; (vii) 鳞翅目昆虫 PCG 密码子碱基组成偏好 AT, 例如 32 种鳞翅目昆虫 A+T 平均含量为 79.4%; (viii) 密码子第 3 位偏爱 AT, 所分析的这 32 个物种 PCG 的 A+T 含量平均值为 92.5%, 而朝灰蝶、天蚕等物种 A+T 含量甚至超过 95%. 另外, 通过与已发表但未被 Codon Usage Database 数据库收录的其他物种如人支蝠蛾和云南蝠蛾[12]、残锷线蛱蝶[16]、O. lunifer[35]、美国白蛾[36]、二化螟和稻纵卷叶螟[41]、小蔗螟[42]等密码子使用的分析数据比较可知, 表 2 中的密码子使用频率与上述数据具有良好的一致性.
(2) 密码子进化. 生物进化模式的差异使得多种密码子系统得以形成: 不仅是细胞核与细胞器(线粒体、叶绿体)之间, 而且细胞核与细胞核、细胞器与细胞器之间的遗传密码也不尽相同[55]. 例如在鳞翅目昆虫线粒体基因组中, 密码子 UGA, AUA, AGA 分别对应 Trp, Met, Ser; 而在通用密码子表中它们分别对应终止子, Ile, Arg, 这 3 个密码子的变异情况也适用于所有节肢动物和绝大部分无脊椎动物[56,57]. AGG 在无脊椎动物线粒体密码子系统中对应 Ser, 而在少数节肢动物线粒体中 AGG 对应 Lys[55]. 不同密码子系统的出现, 可能因为遗传密码进化可以带来直接的选择优势[58]. 影响翻译机器的因素都可能造成遗传密码变异, 其中作用较大的有碱基修饰[59]、 tRNA 突变和 tRNA 编辑[57]. 例如, 在鳞翅目昆虫 mtDNA 中 AGA 对应 Ser 而非 Arg, 这是其反密码子 GCU 经甲基化修饰形成 m7 GCU 所致[57].
1.6 基因间隔区与基因重叠区
A+T 富集区也称控制区或 D-Loop, 是动物 mtDNA 的复制起始区和最大的基因间隔区[43,60]. 除亚洲玉米螟、欧洲玉米螟等 2 个物种(“A+T 富集区” 未确定)外, 其余 56 个物种的 A+T 富集区位置相对固定, 位于 rrnS, trnM(trnI)基因之间. 这 56 种鳞翅目昆虫 A+T 富集区碱基平均含量: A 为 44.1%, G 为 2.2%, C 为 4.3%, T 为 49.4%. 这 56 种鳞翅目昆虫 A+T 富集区的 A+T 含量桑尺蠖最高(98.2%), 天蚕蛾最低 (87.89%), 平均为 93.5%. A+T 富集区大小并不保守 (311~1367 bp, 平均 464 bp), 最小的是大螟, 最大的是人支蝠蛾. 曾有学者指出, A+T 富集区是 mtDNA 进化最快的区域, 可作为动物群体遗传学的重要分子标记, 然而从核苷酸替换的角度而言, A+T 富集区可能并不是昆虫 mtDNA 变化最大的区域[61]. 例如比较家蚕和日本野桑蚕 mtDNA 可知, 它们 A+T 富集区的核苷酸序列变异程度仅高于 rrnS 基因[10]; 比较家蚕和中国野桑蚕也发现 A+T 富集区核苷酸序列差异低于蛋白编码基因 cytb, cox3 和 nad4L[10]. 鳞翅目昆虫 mtDNA 的 A+T 富集区一般存在 4 个保守结构[15,19,23,61], 包括: (1) 始于临近核糖体 RNA 基因一端第 10~30 碱基位点的 ATAGA[20]或 ATAGT 及其后 15~20 bp 的 polyT, 即复制子[61]所在区域. 例如, 至少 35 个物种(35/56)A+T 富集区序列中存在“ATAGA(T)” +“18 bp polyT”元件; 同时, 除人支蝠蛾、云南蝠蛾、华西琉璃灰蝶、金凤蝶、绢粉蝶外, 其余 51 种鳞翅目昆虫(51/56)A+T 富集区均含 ATAGA(T)+polyT 片段. (2) 微卫星元件(AT)n [23]或(TA)n, 靠近转运 RNA 基因一端 . 然 而 , 仅 22 个鳞翅目昆虫物种 (22/56)A+T 富集区含有微卫星片段“(AT)n”(n≥8), 仅 25 个鳞翅目昆虫物种(25/56)A+T 富集区含有“(TA)n” (n≥8), 因此 A+T 富集区微卫星元件的保守性有待进一步研究. (3) 位于转运 RNA 基因上游的 polyA[15,23] 或 polyT, 长约 5~15 bp. (4) 贯穿整个 A+T 富集区的重复序列[15,23], 其大小、重复数目均不保守.
相关知识推荐:基因组测序研究论文发表sci期刊难吗
除 A+T 富集区外, mtDNA 上散在分布着数目不等(1~20 个)、大小不定(几碱基对到几百碱基对)的间隔序列. 线粒体进化趋向于基因组大小减少, 是通过消除/削减基因间隔区(intergenic spacer)实现的, 这一观点已被证实[62,63]. 为探讨基因间隔区和基因重叠区(overlapping region)保守特征, 对 58 种鳞翅目昆虫 mtDNA 中 trnQ—nad2, trnS2—nad1, trnW—trnC, atp8—atp6 等 4 处区域进行统计分析. 统计方法: 登录 GenBank 数据库, 检索并计算 58 种鳞翅目昆虫在此 4 处区域的碱基个数, 统计结果见表 3.
统计结果表明, 鳞翅目昆虫 mtDNA 至少有 2 个相对保守的基因间隔区和 2 个相对保守的基因重叠区. (1) 基因间隔区. 间隔区 1(Spacer1)较大, 处在 trnQ—nad2 基因间, 大小 40~90 bp, 平均 53 bp; 间隔区 2(Spacer2)较小, 处在 trnS2—nad1 基因间, 大小 5~40 bp, 平均 17 bp. 相比于间隔区 2, 间隔区 1 更为保守. 早期研究[16,23,39]通过比对间隔区 1与 nad2基因核苷酸序列发现两者相似性较高, 因此推断间隔区 1 可能来自 nad2 基因. 间隔区 2 一般含有保守序列 ATACTAA[36]. (2) 基因重叠区. 2 个保守的重叠区, 即 atp8—atp6 之间 7 bp 的 ATGATAA 序列(Region1) 和 trnW— trnC 之 间 8 bp 的 AAGCCTTA 序 列 (Region2), 同时存在于绝大多数已知的鳞翅目线粒体基因组参考序列. Region1 位于蛋白编码基因, 而 Region2 序列位于 tRNA 基因, 因此根据 Knibbe 等人[64]的研究,可以推测 Region1 可能比 Region2 更为保守. 对序列变异情况的统计结果(表 3)表明, Region2(7/58)比 Region1 (2/58)变异更大, 证实了上述推测. (3) 与基因间隔区相比, 基因重叠区更为保守[12]. 综上, 从碱基个数来看, 不同区域的保守程度为: 间隔区一致的. (3) 日本野桑蚕和中国野桑蚕何时发生的分歧? Yukuhiro 等人[13]、潘敏慧等人[82]、孙伟等人[83] 发现两者线粒体基因组序列差异较大, 并分别根据线粒体基因 nad5, cox1/cox2, cox1/A+T 富集区核苷酸替换速率, 计算得出两者分歧时间分别为 7.1 Mya (million years ago), 0.95~1.66 Mya 和 23600 Ya(years ago). (4) 群体遗传学研究. Li 等人[77]通过比较分析 41 个蚕类(家蚕和野桑蚕)线粒体基因组序列, 揭示了家蚕驯化过程中群体结构的变化, 认为家蚕和中国野桑蚕群体大小均未经历扩张或收缩. 然而, 这一推断与最近基于线粒体基因和核基因联合分析得出的家蚕在约 1000 年前经历群体扩张[83]这一推断并不一致. 总之, 线粒体基因组序列在鳞翅目昆虫物种起源和进化研究中发挥了重要作用; 对于一些有争议的问题, 增加核基因序列数据可能获得更全面的认识.
2.3 性别控制
沃尔巴克氏体(Wolbachia)是广泛存在于昆虫体内的一种内共生细菌, 能影响宿主的生殖行为, 进而控制宿主性别比例[72]. 已发现 Wolbachia 可影响 mtDNA 核苷酸序列(即降低或增加 mtDNA 多态性) [72], 调控线粒体蛋白编码基因的表达[84], 并最终导致孤雌生殖和雄性致死等 [85]. 例如在幻紫斑蛱蝶 (Hypolimnas bolina) [86], Wolbachia 通过提升被感染雌性个体的相对适合度(relative fitness)而快速传播, 进而快速提升雌蝶数量; 在小菜蛾(Plutella xylostella), 未感染 Wolbachia 种群雌雄比例为 1:1, 感 染 Wolbachia 种群雌雄比例为 2:1[87]. 关于 Wolbachia 影响 mtDNA 多态性的具体分子机制, 最近的实验证据表明, 两者之间不存在直接的相互作用关系: 在侵入宿主过程中, Wolbachia 与线粒体的共同传递使得 Wolbachia 能够影响 mtDNA 多态性, 且这种影响是一种间接效应[88]. 但是, Wolbachia 通过哪些途径发挥作用, 以及 Wolbachia 作用于哪些 mtDNA 位点均有待进一步的研究.
2.4 其他
(1) 线粒体 DNA 复制. 线粒体 DNA 的复制起始点(replication region, OR)位于 A+T 富集区, 且主链、次链各有一个独立的 OR. Saito 等人[89]研究发现, 尽管全变态昆虫(包括鳞翅目昆虫)OR 具体位置不同, 但它们都有一个共同的特征, 即位于长约 10~20 bp 的 polyT 之后, 因此认为 polyT 可能参与了全变态昆虫 mtDNA OR 的识别. 由于 Saito 等人[89]的实验以果蝇为主, 鳞翅目昆虫仅分析了家蚕, 因此其研究结论是否同样适用于其他鳞翅目物种还有待检验. 本文以序列(T)10 搜索 56 种鳞翅目昆虫 A+T 富集区, 发现除旋纹潜蛾外, 其余 55 个物种在靠近 rrnS 基因一端均存在 10 bp 及以上的 polyT, 因而基本支持“全变态昆虫 mtDNA 的 OR 位于 polyT 下游” 这一结论.
(2) 碱基偏好的形成机制. 鳞翅目线粒体基因组碱基组成表现明显的 AT 偏好, 其形成机制是什么? 一般认为可能与线粒体起源进化有关[90], 而线粒体由单源的内共生细菌进化而来[91], 提示可以从其他内共生细菌获得间接证据. 例如, Buchnera aphidicola 是蚜虫(aphids)体内的一种内共生细菌, 基因组富含 AT. Wernegreen 等人[92]通过分析发现, 在与宿主漫长的共生过程中, Buchnera 基因组高 AT 含量可能由于其高 AT 突变及长期遗传漂变导致. 这些信息对于探讨鳞翅目线粒体基因组的 AT 偏好具有借鉴意义.
3 结语与展望
鳞翅目作为昆虫纲第二大目, 不但物种资源丰富, 而且对人类社会影响深远. 相比鳞翅目庞大的物种数, 目前已完成线粒体全基因组测序的鳞翅目种类还比较少, 因此, 获得更多鳞翅目物种的线粒体基因组序列显得颇为迫切. 在利用各种测序方法[93]获取线粒体基因组序列的基础上, 研究者可进行后续的数据库与网站建设、线粒体基因组注释、线粒体比较基因组学、线粒体转录组学、线粒体蛋白质组学等研究. 在应用研究方面, 关于鳞翅目高级阶元系统发育关系如“大鳞翅类不同总科之间的关系”、“凤蝶总科不同科之间的关系”等研究还存在不少争议, 线粒体基因组序列的测定有望为解决争议问题提供新的途径和手段. 最后, 基于线粒体基因组研究鳞翅目物种的起源与进化、Wolbachia, mtDNA 和鳞翅目宿主性别比例三者的具体关系等, 将有望为鳞翅目农林害虫防治、经济昆虫种质资源的利用等提供新的思路和方法.——论文作者:王维①② , 孟智启① , 石放雄② , 李风波①*