发布时间:所属分类:农业论文浏览:1次
摘 要: 摘要质子交换膜燃料电池作为新型绿色能源技术,在未来电动汽车及分散式电站等领域将有非常广阔的应用前景。但目前制约燃料电池商业化的主要瓶颈是其高成本和低寿命,而贵金属氧还原催化剂的使用是成本的主要来源,也是决定燃料电池性能的关键因素。本文综述
摘要质子交换膜燃料电池作为新型绿色能源技术,在未来电动汽车及分散式电站等领域将有非常广阔的应用前景。但目前制约燃料电池商业化的主要瓶颈是其高成本和低寿命,而贵金属氧还原催化剂的使用是成本的主要来源,也是决定燃料电池性能的关键因素。本文综述了一种新型的氧还原催化剂——铂基金属间化合物,介绍其结构特性以及过渡金属组成对氧还原催化反应活性和稳定性的影响,最后对铂基催化剂的发展给出了展望。
关键词质子交换膜燃料电池,氧还原反应,电催化剂,金属间化合物
1.引言
在全球能源危机以及环境问题不断恶化的严峻形势下,发展清洁与可再生能源已成为近年来世界各国关心的重要议题。在各类可持续发展的新能源中,氢能因具有出色的比能量密度优势及广泛的技术适应性,被认为是未来最具有发展前景的新能源之一。质子交换膜燃料电池(ProtonExchangeMembraneFuelCells,PEMFCs)技术,将氢和氧中的化学能直接转化为电能,是氢能利用最直接有效的方式[1]。近五十年,PEMFCs技术已得到迅速发展,目前最为广泛的应用就是作为电动汽车动力源,2014年日本丰田公司量产并商业化的燃料电汽车(FuelCellVehicle,FCV)Mirai标志着燃料电池商业化时代的到来[2]。
在燃料电池实际应用中,阴极氧还原反应(OxygenReductionReaction,ORR)是一个动力学慢反应,其反应速率直接决定了电池的性能,需要使用贵金属Pt作为催化剂[3]。然而Pt为稀有金属,全球储量有限,价格昂贵,致使目前PEMFCs面临成本高的问题。另一方面,目前广泛使用的Pt/C催化剂在长时间工作条件下存在纳米颗粒团聚、溶解、再沉积以及脱落的问题,使催化剂性能显著下降,电池稳定性能不好。为了降低催化剂中Pt的用量,近年来国内外的研究热点为Pt与过渡金属M(M=Fe,Co,Ni,Cu,Pd等)形成的合金催化剂,目前已有许多合金催化剂的相关报导和综述性文章[3][4][5],对其现状和发展方向给予了评论。但是,在燃料电池阴极富氧和高电压的酸性条件下,合金催化剂中的过渡金属存在易溶出,并污染质子交换膜的问题,使电池性能迅速衰减。近年来,一种新型的ORR催化剂——具有有序结构的金属间化合物,因其表现出更高的催化活性和稳定性,吸引了国内外研究工作者的广泛关注。本文将根据近些年对于这类催化剂的研究,从催化剂的组成、结构、性能三个角度出发,综述几类典型的金属间化合物ORR催化剂的研究进展,并对其面临的挑战和发展方向给出一些展望。
2.金属间化合物的有序结构与催化活性
对于纯铂催化剂的研究及ORR反应机理密度泛函理论(DensityFunctionalTheory,DFT)计算表明,有效氧还原催化剂表面的电子状态应既有利于氧气分子的吸附并促使O=O双键断裂,又有利于反应中间产物从催化剂表面脱离,使反应顺利进行。Nørskov等人[6]的计算显示,纯金属铂表面对于氧分子的结合能过于强,应对铂催化剂进行合理调控,使其表面对氧气的吸附能比Pt{111}晶面弱约0.2eV,以提高其催化活性。基于这样的理论,PtM合金催化剂被提出并广泛研究,其对于ORR催化活性的提高主要表现在两个方面:几何效应和电子效应。一方面,由于过渡金属原子直径比铂原子小,合金化作用使原有的铂-铂原子间距收缩(几何效应);另一方面,由于过渡金属更容易失电子,使合金颗粒表面电子结构发生改变(电子效应)。两者的协同作用有利于氧气分子在颗粒表面的双址吸附,以及反应产物的脱附,从而进一步提高催化活性[7]。但是,在这类合金中,过渡金属原子以固溶体形式无序的掺杂在铂晶格中,原子排列仍保持原有铂的面心立方结构(face-centeredcubic,fcc),合金催化剂所产生的几何效应和电子效应都不均匀,因而催化活性的提高也有限。同时,无序的过渡金属原子排列使其在酸性条件下很容易从合金颗粒中溶出,存在长时间循环过程中过渡金属溶解的问题,使催化性能迅速下降。
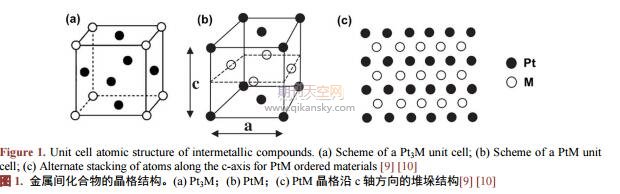
金属间化合物是由两种或多种金属组元按比例结合形成具有不同于其它组成元素的长程有序晶体结构,并且具有金属基本特性的化合物。与合金结构不同,在金属间化合物的晶格结构中,Pt和M原子都有序的占据晶格中相应格点,并以金属键或离子键相互作用,使整个晶体呈现出长程有序的晶系结构,这种长程有序的结构使其具有特殊的催化特性[8]。根据不同原子比例,一般金属间化合物分为Pt3M,PtM,PtM3相,如图1所示。在Pt3M中(图1(a)),Pt原子占据正方体晶格六个面的中心位置,而M原子占据正方体的八个顶点(PtM3相反);而在PtM中,Pt与M按原子层交替排列(图1(b)~(c)),这种原子排列具有面心四方结构(facecenteredtetragonal,fct)[9][10]。这种长程有序的结构不仅使过渡金属的调控作用得到充分发挥,而且与铂原子形成的化学键能更加稳定过渡金属原子,使其不易在酸性条件下溶解,进而更进一步的提高了催化活性和稳定性。近几年对PtM金属间化合物的研究结果均表明具有有序结构的金属间化合物催化剂比无序结构的合金催化剂表现出更好的催化活性和稳定性。早在2004年,Abruña[11]研究小组就报道了有序金属间化合物电催化剂在燃料电池中的应用,他们制备了一系列的Pt基金属间化合物发现这些催化剂表现出比铂合金和纯金属铂更高的催化活性。
3.Pt基金属间化合物
3.1.PtFe金属间化合物
金属间化合物因具有特殊的有序结构因而表现出优异的物理和化学性能,其早期应用主要是作为超高密度磁性存储材料而吸引了广泛的关注。ShouhengSun等人于2000年报道了单分散PtFe纳米颗粒的制备方法,在油酸和油胺混合溶液中,他们采用化学还原乙酰丙酮铂(Pt(acac)2)和热分羰基铁(Fe(CO)5)的方法,得到了无序结构的PtFe纳米颗粒。经过500℃的热处理,无序结构转变为有序结构的fct-PtFe纳米粒子[12]。他们继续研究了fcc-PtFe和fct-PtFe纳米颗粒在0.5MH2SO4中的阴极氧还原催化活性,发现有序结构的PtFe纳米颗粒表现出比无序颗粒更高的催化活性,并且在酸性溶液中,有序结构中Fe含量降低只有3.3%,而无序结构中Fe含量的损失达到36.5%,充分说明了有序结构对于铁原子的稳定作用[13]。为了提高催化效率和贵金属的利用率,催化剂材料一般要求具有最大的表面积与体积比,即较小的颗粒尺寸以增加其比表面积,提高贵金属的利用率[14]。但高温热处理过程往往会引起纳米颗粒的团聚长大,使催化剂的初始活性下降。在接下来的研究中,他们在热处理的过程中,采用MgO包覆PtFe-Fe3O4颗粒,得到完全有序结构的fct-PtFe纳米颗粒。这一催化剂在ORR极化曲线测试中表现出0.958V的半波电位,其在0.9V的比表面积比活性和质量比活性分别达到3.16mA/cm2和0.69A/mgPt,明显优于部分有序结构和纯铂催化剂[15]。
另一方面,在制备过程中采用碳层保护也是阻止纳米颗粒长大的一种方法。XinxinDu等人采用化学气相沉积法,以Fe(CO)5为催化剂、C2H2为碳源先制备得到Fe/C混合物,再采用液相化学还原法,在所得Fe/C混合物表面沉积铂纳米颗粒。在还原过程中,部分铁团簇被氧化,与铂离子发生置换反应,因而形成PtFe颗粒被非晶碳层包覆的结构,在接下来的热处理过程中直接转化为有序的PtFe纳米颗粒,其平均直径约为3.6纳米。所得的催化剂在0.1MHClO4溶液中的半波电位达到0.89V,经过3000个电位循环后,半波电位下降了59mV,但Pt/C催化剂的半波电位下降了140mV,充分说明了有序化合物的催化稳定性得到提高[16]。DongYoungChung等人报道了采用聚多巴胺包覆fcc-PtFe纳米颗粒,然后在约700℃条件下热处理,得到有序的fct-PtFe催化剂。在这一过程中,聚多巴胺原位碳化,直接形成氮掺杂的碳层,包覆在纳米颗粒表面,有效的阻止其团聚长大。在0.9V,其比表面积比活性和质量比活性分别为2.3mA/cm2和1.6A/mgPt,而且,在电位循环的寿命测试中,该催化剂表现出很好的稳定性,几乎没有性能的衰减[17]。ChanwonJung等人采用液相还原的方法先制备了Pt-Fe合金纳米颗粒,将其负载在碳载体上后,用1,2十六烷二醇和乙酰丙酮铁为碳源在其表面形成包覆层,经过一系列热处理后得到均匀分散的有序金属间化合物Pt3Fe/C纳米颗粒。该催化剂的质量比活性达到0.454A/mgPt,由于碳层的保护作用,在长时间的循环过程中没有发生颗粒的团聚和铁的溶出。DFT理论计算表明,铁原子在有序金属间结构中具有比合金中更高的溶解电位,因而比合金Pt-Fe/C更好的稳定性[18]。
随着制备工艺的不断改进,目前采用多元醇还原的方法,已不需要在合金颗粒表面包覆,即能获得相对较小并均匀分散的金属间纳米颗粒。蔡业政等人[19]报道了改进多元醇方法制备碳载铂铁合金催化剂前驱体,然后在惰性气体环境中煅烧,将无序结构合金转化为有序结构的铂铁金属间化合物催化剂。所得催化剂颗粒的尺寸分布为4nm~6nm,且均匀负载在载体上,电化学测试结果显示,其催化活性均高于商业Pt/C催化剂。虽然PtFe金属间化合物纳米颗粒表现出了更高的催化活性和稳定性,但是实际燃料电池运行过程中,特别是在电池起停及高电位运行条件下,还是会有少量铁溶解出来。溶出的铁以离子状态存在,与极少量的氧还原反应副产物H2O2即可反应生成具有强氧化性的自由基(即芬顿试剂)。这些自由基会攻击质子交换膜和固体聚合物,使其发生氧化分解,导致质子交换膜破裂,电池失效。因而,目前国际上已经很少使用含有铁基的合金或金属间化合物作为氧还原催化剂。
3.2.PtCo金属间纳米结构
一般而言,由于只有催化剂颗粒表面原子才能接触到电解液和反应物,电催化反应主要发生在纳米颗粒表面,颗粒内部的贵金属不能参加催化反应,造成贵金属利用率低下。为了进一步提高催化活性和铂的利用率,研究人员开发了具有核壳结构的纳米催化剂,通常以低价格金属作为核,多层铂原子为壳层。核的组成和壳层的形貌及二者的相互作用将对催化活性产生至关重要的影响,理论计算研究预测Pt基双金属核壳结构催化剂将具有较好的催化活性和稳定性[20]。钴元素是另一种极易与铂形成金属间化合物,并且对铂催化活性提高有很大帮助的过渡金属元素。通过精确调控核的结构和组成,可以获得以PtCo有序金属间化合物为核的纳米颗粒[21]。DeliWang等人[22]报道了有序结构的Pt3Co@Pt/C催化剂,他们将氯铂酸(H2PtCl6·6H2O)和氯化钴(CoCl2·6H2O)溶解在水中,再将碳黑载体分散在上述溶液里,加热蒸发掉溶剂并干燥后,将研磨过的粉末在H2/N2混合气体中分两个阶段加热,最终得到有序的核-壳结构催化剂。实验结果表明,热处理温度达到700℃时,无序结构才能完全转变为有序结构,并且颗粒表面由2~3层Pt原子的排列。在0.1MHClO4电解液中,该催化剂的比表面积比活性和质量比活性分别是Pt/C催化剂3倍和9倍。YezhengCai等人[23]用多元醇还原方法先得到PtCo合金纳米颗粒,对比不同热处理温度,发现700℃时制备的Pt3Co/C-700和PtCo/C-700表现出最好的催化活性,充分说明温度是影响有序化的重要因素。
推荐阅读:化工实验论文投稿指导
获得核–壳结构催化剂的另外一种方法是去合金化,即通过化学或电化学浸出的方式选择性去除合金纳米颗粒表面的过渡金属,再经过热处理使表面Pt原子重排,形成致密的壳层结构[24]。JunruiLi等人[25]采用液相共还原法先制备出CoPt纳米颗粒,沉积到碳载体上后进行第一次热处理,得到具有有序结构的L10-CoPt/C纳米颗粒。再将该颗粒在0.1MHClO4溶液中60℃浸泡24小时,分离干燥后在95%Ar和5%H2混合气体下400℃热处理2小时,最终得到L10-CoPt@Pt/C核—壳结构催化剂。这一催化剂的比表面积比活性和质量比活性分别达到8.26mA/cm2和2.26A/mgPt,分别是商业Pt/C的38倍和19倍。为了模拟该催化剂在实际电池中的稳定性,加速老化实验在60℃的条件下进行,经过30,000个循环后,L10-CoPt@Pt/C的极化曲线几乎没有发生变化,表现了非常好的稳定性。在实际燃料电池测试中,0.9V时催化剂的质量比活性为0.56A/mgPt,30000个循环后只有19%的降低。通过DFT理论计算对比不同催化剂表面铂层自由能的结果表明,当铂原子层厚度相同时,以CoPt为核的催化剂表面对氧还原反应中间产物的吸附能被削弱,证明可以通过应力作用和配位作用对催化剂表面进行有效调控,使其更有利于氧还原反应的发生。计算结果同时证实了Co原子对表面应力的改变作用强于Fe原子,这也是L10-CoPt@Pt/C催化剂性能优于PtFe催化剂的主要原因。这种以PtCo金属间化合物为核,多层铂原子为壳的核壳催化剂具有最大的潜力,成为替代Pt/C的阴极氧还原催化剂。